
Plaquetas:
Para hablar de la hemostasia debemos hablar en
primer lugar de las plaquetas. Estas provienen de la unidad formadora de las
colonias del bazo y esta unidad da lugar a un precursor ya diferenciado que es
la unidad formadora de colonias de megacariocitos que dará lugar a las
plaquetas. La primera línea de la unidad formadora de megacariocitos se divide,
se diferencia y da lugar a un megacarioblasto. En esta fase de megacarioblasto
sintetiza ADN hasta alcanzar una poliploídia de 8 a 32 núcleos. Por lo tanto estas
células son células gigantes en donde la síntesis de ADN lo lleva a la división
citoplasmática. Lo que ocurre es una endomitosis.
 |
| Plaquetas vistas a ME |
Síntesis
de plaquetas:
La síntesis de los elementos constitutivos de las
plaquetas comienza en la síntesis de megacarioblastos. En las etapas
posteriores dejó de sintetizarse ADN, simplemente la célula sufre procesos de
maduración mediante los cuales los núcleos se lobulan, se condensa la
cromatina, en el citoplasma aparecen abundantes granulaciones lisosómicas y al
mismo tiempo la membrana citoplasmática se invagina, desarrollándose otras
membranas llamadas membranas de demarcación. Estas membranas fraccionan el
citoplasma y se unen y rodean zonas de citoplasma con gránulos que serán las
futuras plaquetas.
En el microscopio se ven como puntitos y tienen un
diámetro de 3 o 4 micras y constan de una estructura que se llama cronómero
rodeado de una zona o lado claro que se llama hialómero. No tienen núcleo y
tienen forma de discos redondos u ovalados y sus granulaciones contienen varias
substancias para la coagulación. Su vida media es de ocho días y su número está
comprendido entre 250.000 a 300.000 mil por cm3. Pero este número
puede variar por ejemplo con la altitud, que disminuye el número de plaquetas o
con la estación del año.
Hemostasia:
Nos referimos a todas las reacciones que ocurren en
nuestro organismo para impedir o minimizar la pérdida de sangre cuando se
produce la rotura de un vaso. En la hemostasia se llevan a cabo cuatro tipos de
reacciones (en tiempo de hemostasia). Estos cuatro tiempos pueden ser
simultáneos.
1. La primera es el tiempo de hemostasia vascular o
de vasoconstricción.
2. El segundo tiempo de hemostasia es el tiempo
plaquetario.
3. El tercer tiempo es el tiempo plasmático o de
coagulación (sin plaquetas no se coagula pero las plaquetas no coagulan).
4. El cuarto tiempo es de de fibrinólisis.

1. Tiempo vascular o de vasoconstricción:
La lesión de un vaso sanguíneo provoca una
respuesta contráctil por lo tanto la vasoconstricción del músculo liso del vaso
y el estrechamiento de la luz del vaso. La vasoconstricción es un mecanismo
reflejo que se debe fundamentalmente al estímulo de las fibras nerviosas
simpáticas que inerven el músculo liso de ese vaso.

2. Tiempo plaquetario:
Entonces tenemos el segundo tiempo que es el tiempo
plaquetario. Este tiempo tiene varias etapas que son la etapa de adhesión, la
etapa de activación-secrección y la etapa de agregación. Estas etapas conducen
a la formación del tapón plaquetario y junto con el primer tiempo se conoce con
el nombre de hemostasia primaria.

Al mismo tiempo que se agregan se activa un
fosfolípido de la membrana plaquetaria que es el que va a iniciar la coagulación. La agregación está
controlada por una prostaciclina (la prostaciclina PGI2). Esta PGI2
inhibe la coagulación, se forma en el metabolismo del ácido araquidónico en el
endotelio vascular y tiene acción vasodilatadora y antiagregante plaquetario.
Por lo tanto en nuestro organismo hay un equilibrio entre vasoconstricción y
vasodilatación y entre agregación y antiagregación. Este equilibrio está controlado
por dos substancias: el tromboxano A2 plaquetario y la PGI2
vascular y lo que hace es limitar la formación del tapón de plaquetas a la zona
de la lesión. Si no existiera este equilibrio tendríamos una coagulación
incontrolada sin lesiones o sería imposible coagular aún sin lesiones.
3. Tiempo plasmático o de coagulación:
Se llama también hemostasia secundaria y es un
proceso muy complejo en el cual una proteína plasmática soluble llamada
fibrinógeno se convierte en una insoluble fibrina que es la que forma la red
del coágulo.
 La coagulación se produce por la activación secuencial de una
serie de factores, que son proteínas, que se encuentran en el plasma de forma
inactiva y que se van a activar secuencialmente mediante una reacción en
cascada. Pero la coagulación se produce en tres fases:
La coagulación se produce por la activación secuencial de una
serie de factores, que son proteínas, que se encuentran en el plasma de forma
inactiva y que se van a activar secuencialmente mediante una reacción en
cascada. Pero la coagulación se produce en tres fases:

a. La
primera fase consiste en la formación del activador de la protombina. Esta
formación puede ocurrir por dos vías, por una vía intrínseca o por una vía
extrínseca. En las dos vías se va a llegar a la activación del factor X y a
partir de ahí las dos vías son comunes. Las dos vías se producen normalmente
simultáneamente pero la intrínseca se da cuando la lesión es en la propia
sangre (como por ejemplo una trombosis) y la extrínseca cuando la lesión afecta
también al tejido subadyacente.
La vía intrínseca se
inicia cuando la sangre entra en contacto con la superficie dañada o, en
laboratorio, cuando se encuentra en contacto con la pared de vidrio del tubo de
ensayo. Este contacto de la sangre con la superficie dañada provoca que el
factor XII se active y se transforme en factor XII activado. Este factor XII
activado en presencia de calicreína y quininas actúa sobre el factor XI de
coagulación transformándolo en factor XI activado. Este factor XI activado en
presencia de Ca++ actúa sobre el factor IX transformándolo en factor
IX activado. Este factor IX activado conjuntamente o en presencia de calcio,
fosfolípidos plaquetarios y factor VIII, todo ello actúa sobre el factor X
transformándolo en factor X activado.
La vía extrínseca,
más rápida se inicia cuando el factor VII de coagulación es activado en
presencia de Ca++ por el factor tisular que se ha cuando se ha
producido la rotura del tejido. Este factor VII activado en presencia de Ca++
y fosfolípidos plaquetarios actúa sobre el factor X transformándolo en factor X
activado.
Este factor X activado obtenido por cualquiera de estas
vías forma complejos con el factor V de coagulación más con los fosfolípidos
plaquetarios en presencia de Ca++ , formando un complejo que es el
activador de la protrombina.

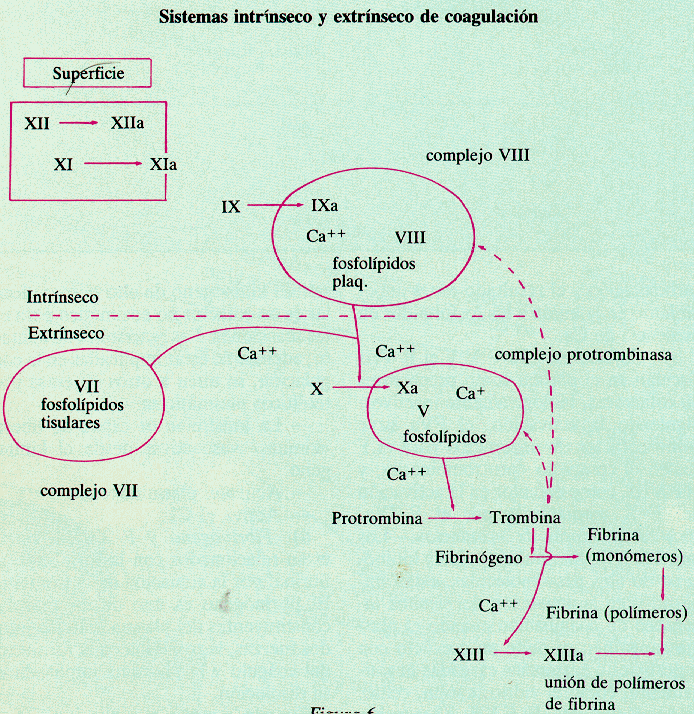
b. Una
vez que tenemos el activador de la protrombina (FV + FXACTIVADO +
Fosfolípidos plaquetarios) entramos en la fase dos que consiste en la
transformación de la protrombina en trombina mediante el activador de la
protrombina
c. Tenemos
la trombina y entramos en la fase tres. En esta fase la trombina actúa sobre la
proteína plasmática fibrinógeno y produce roturas en su estructura formando
monómeros de fibrina pero siempre para que actúe tiene que haber Ca++.
Pero al mismo tiempo la trombina convierte el factor XIII en factor XIII
activado y este factor XIII activado, que también se llama factor estabilizante
de la fibrina, en presencia de calcio provoca que se ensamblen los monómeros de
fibrina y formen unos polímeros de fibrina que son insolubles que forman la red
del coágulo, que es insoluble.
La mayor parte de
los factores de coagulación se sintetizan en el hígado y es necesario de la
vitamina K en la síntesis de muchos de
ellos (a los recién nacido les ponen vitamina K). Una vez formado el coágulo,
la actina y la miosina de las plaquetas atrapadas en la red del coágulo
reaccionan entre ellas y se contraen y, por lo tanto, hay una contracción
resultante de las fibras de fibrina hacia las plaquetas y por lo tanto
disminuyen las plaquetas del coágulo, aproxima los bordes de la lesión
facilitando la reparación. Este proceso lo inicia la trombina al inducir la
liberación del calcio almacenado en el citoplasma de las plaquetas. Por lo
tanto entramos en el cuarto tiempo.
4. Tiempo de fibrinólisis.
El cuarto tiempo o tiempo de fibrinólisis supone la
lisis del coágulo, de la fibrina. A medida que prosigue la reparación del
tejido dañado, la reparación tisular, el coágulo se disuelve gradualmente. Pero
para que esto se logre un activador transforma un componente plasmático llamado
plasminógeno o profibrinolina en plasmina o fibrinolisina, y esta plasmina o
fibrinolisina es quien disuelve el coágulo. Esta es una enzima proteolítica
(rompe los enlaces de las proteínas, los enlaces peptídicos) parecida a la
tripsina del aparato digestivo y lo que hace es digerir los anillos de fibrina
pero también digiere substancias de la sangre vecina al coágulo como
fibrinógeno, factor V, factor VIII, factor XII y protrombina. De este modo
evita que se vuelva a iniciar la coagulación en esa zona mientras esta no está
recuperada totalmente.

FACTOR VIII FACTOR ANTIHEMOFÍLICO
FACTOR XIII FACTOR ESTABILIZANTE DE LA FIBRINA
El 83% de los
hemofílicos tienen hemofilia clásica, debida a la carencia de factor VIII, el
15% carecen de factor IX y el 2% de factor XI. Los dos primeros son una
enfermedad recesiva ligada al sexo (a X), mientras que la tercera es
autosómica. La familia real española tiene la hemofilia clásica al igual que la
mayoría de las familias europeas.
Prevención de la coagulación
inadecuada mediante coagulantes endógenos:
Las células endoteliales
vasculares no lesionadas previenen la coagulación liberando substancias que la
inhiben y que llamamos anticoagulantes. Estos anticoagulantes endógenos son los
siguientes:
En primer
lugar tenemos las prostaciclinas (PG). La PG es un poderoso inhibidor de la
agregación plaquetaria.
En segundo lugar tenemos en nuestro organismo la trombomodulina que es
producida en nuestro organismo por las células endoteliales normales y es una
proteína que se une a la trombina formando un complejo, el trombomodulina-trombina
que activa a la proteína C. Esta proteína C es la que va a inhibir al factor V
y al factor VI activado pero además estimula la producción de la enzima
plasmina a partir del plasminógeno.
En tercer lugar tenemos la heparina que es un proteoglucano de carga
negativa que está presente en el plasma (posiblemente a partir de los basófilos
y otras células) y también está presente en la superficie de las células
endoteliales de los vasos sanguíneos. La heparina es soluble en agua pero poco
soluble en líquidos orgánicos y su máximo poder anticoagulante es a 37oC.
La heparina, debido a las cargas negativas reacciona fácilmente con las
proteínas por eso la heparina orgánica tiene que suministrarse en vena y,
además, su vida media es corta, de unas cuatro horas por lo que una vez que se
libera tiene un poder anticoagulante de
cuatro horas.
Por eso se han desarrollado una serie de
investigaciones para obtener anticoagulantes sintéticos. Los anticoagulantes
sintéticos más utilizados son las heparinas sintéticas protegidas, que no
tienen cargas negativas expuestas para que no reaccionen y además se suelen
utilizar anticoagulantes sintéticos derivados de las cumarinas (el sintrom). La
heparina actúa a diversos niveles.
a. En primer
lugar disminuye la adhesividad de las plaquetas.
b. En segundo
lugar impide la transformación de protombina en trombina, no deja actuar al
activador de la protrombina.
c. En tercer
lugar no deja actuar a la trombina.
d. En cuarto
lugar impide la polimerización de los monómeros de fibrina.
e. En quinto
lugar lisa o rompe un coágulo ya formado.Por lo tanto, en ese aspecto,
tiene el mismo efecto que la fibrino-lisin
Imprimir
No hay comentarios:
Publicar un comentario